CUT&RUN
CUT&RUN stands for cleavage under targets and release using nuclease. The technique developed from the Henikoff Lab offers a new approach to pursue epigenetics. CUT&RUN introduces some major modifications to eliminate shortcomings inherent to ChIP-seq. It is simple to perform and inherently robust, with extremely low backgrounds requiring only ~1/10th the sequencing depth as ChIP. CUT&RUN is cost-effective for transcription factor and chromatin profiling.
You are new to CUT&RUN? Learn more about this application in the following sections or jump directly to our webinar series covering CUT&RUN and CUT&Tag.
CUT&RUN Antibodies
A range of antibodies validated for use in CUT&RUN assays.
CUT&RUN Sets
CUT&RUN made easy - with our sets & components
CUT&RUN ConA Beads
Magnetic ConA Beads for usage in CUT&RUN assays
CUT&RUN - Better than ChIP-seq!
CUT&RUN Workflow: Extraction Without Fragmentation
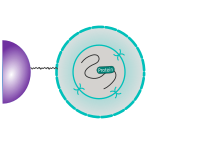
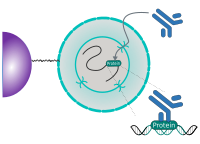
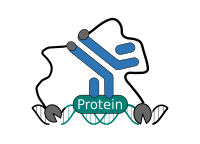
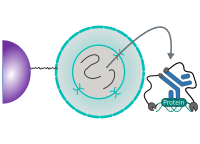
CUT&RUN is performed in situ on immobilized, intact cells without crosslinking. DNA fragmentation is achieved using micrococcal nuclease fused to Protein A and/or Protein G (pAG-MNase). The fusion protein is directed to the desired target through binding of the Protein A/G moiety to the Fc region of an antibody bound to the target. DNA under the target is subsequently cleaved and released and the pAG-MNase-antibody-chromatin complex is free to diffuse out of the cell. DNA cleavage products are extracted and then processed by next generation sequencing (NGS).
The standard CUT&RUN protocol is primarily intended for profiling of histone proteins and transcription factors that bind to chromatin. Reliable detection of rare transcription factors, transient interactions, or proteins in complexes that are not directly associated with DNA have proven difficult using CUT&RUN on uncrosslinked samples. Preserving protein-protein and DNA-protein interactions using crosslinkers on the other hand can introduce artifacts. The following depiction shows the standard CUT&RUN protocol:
CUT&RUN Low Volume and Urea (CUT&RUN Lov-U) is a modification of the original procedure aimed at difficult to characterize interactions in situ while avoiding crosslinking. Initial nuclear extraction minimizes sequestration of the primary antibody and pAG-MNase by non-not chromatin-bound targets in the cytosol. Low volumes throughout the protocol facilitate parallel processing of samples which benefits scalability and reprodcibility. Lastly, urea is used as chaotropic reagent to denature samples in situ and release CUT&RUN products, followed by DNA clean-up on beads.
One of the drawbacks of CUT&RUN is the need to end-polish and ligate sequencing adapter prior to the preparation of a sequencing library. A combination of the CUT&RUN protocol and tagmentation by a hyperactive Tn5 transposase resulted in the CUT&Tag method. Cells are immobilized using magnetic Concanavalin A beads and reversibly permeabilized using digitonin. Instead of the directed nuclease cleavage however, DNA is fragmented by a pA/G-Tn5 loaded with sequencing adapter duplexes. Sequencing adapters are attached to the DNA fragments directly during tagmentation. No further DNA end processing is necessary and the fragments can be used for sequencing library preparation. In addition, CUT&Tag is inherently less sensitive to endogenous DNA damage than CUT&RUN because the transposition takes place on intact double stranded DNA molecules.
Learn more about CUT&Tag: Workflow, Advantages, Products
CUT&RUN Antibodies
A range of antibodies validated for use in CUT&RUN assays.
CUT&RUN Sets
CUT&RUN made easy - with our sets & components
CUT&RUN ConA Beads
Magnetic ConA Beads for usage in CUT&RUN assays
Discover our CUT&RUN Antibodies!
Show moreCUT&RUN and CUT&Tag Target Antigens
Histone modifications such as the repressive H3K27me3 or H3K4me3 associated with active promoters are commonly used antigens for both CUT&RUN and CUT&Tag. Additional antigens include transcription regulators like transcription factors CTCF or SOX2, chromatin modifiers like HDAC2, or proteins involved in nucleic acid metabolism such as Pol II. G-quadruplexes and m6A are among non-protein targets. Tags like e.g. FLAG, HA, or GFP are options for recombinantly expressed target proteins.