KCNQ4 Antibodies
Your search for reliable KCNQ4 antibodies ends here. KCNQ4, known by aliases such as KCNQ4, kcnq4, Kcnq4, is an integral part of our antibody range. Whether you're working with Human, Mouse, Rat, Rabbit, Bat, or other species, our range of KCNQ4 antibodies offer precise detection across diverse samples. These specialized antibodies are tailored for various scientific applications like WB, IHC, IF, ICC, ELISA, providing you with options like polyclonal, recombinant, and monoclonal antibodies, sourced from different host species such as Rabbit, Mouse, Goat. The efficacy of our antibodies is well-established, demonstrated through multiple methods.
Detailed information, including references, images, and validations by other customers, can be found on each product page. Should you require assistance in finding a specific product, our customer service team is ready to assist. Utilize our KCNQ4 antibodies in your research endeavors for dependable KCNQ4 detection.
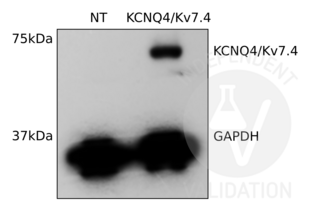
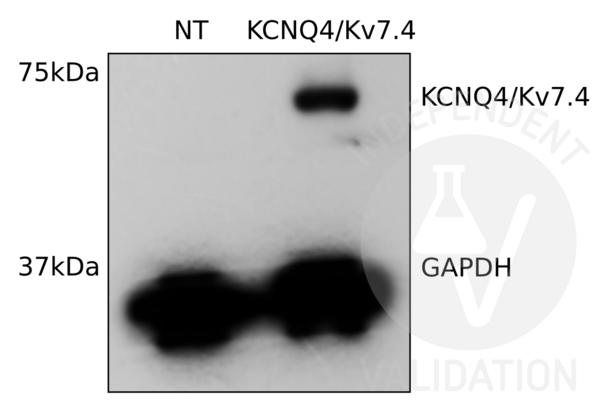 KCNQ4 antibody (C-Term) (ABIN3185286)
KCNQ4 antibody (C-Term) (ABIN3185286)
KCNQ4 Reactivity: Human, Mouse WB, ELISA Host: Rabbit Polyclonal unconjugated
KCNQ4 Reactivity: Human WB, IHC, IP, IF, ICC, AA Host: Mouse Monoclonal N43-6 (Formerly S43-6) unconjugated
KCNQ4 Reactivity: Human WB, IHC, IP, IF, ICC, AA Host: Mouse Monoclonal N43-6 (Formerly S43-6) HRP
KCNQ4 Antibodies by Grade
Find KCNQ4 Antibodies with a specific Grade. The Grade listed below are among those available. Click on a link to go to the corresponding products.
KCNQ4 Antibodies by Reactivity
Find KCNQ4 Antibodies for a variety of species such as anti-Human KCNQ4, anti-Mouse KCNQ4, anti-Rat KCNQ4. The species listed below are among those available. Click on a link to go to the corresponding products.
KCNQ4 Antibodies by Host
Find KCNQ4 Antibodies with a specific Host. The Host listed below are among those available. Click on a link to go to the corresponding products.
KCNQ4 Antibodies by Clonality
Find available monoclonal or polyclonal KCNQ4 Antibodies. Click on a link to go to the corresponding products.
KCNQ4 Antibodies by Clone
Find KCNQ4 Antibodies with a specific Clone. The Clone listed below are among those available. Click on a link to go to the corresponding products.
Popular KCNQ4 Antibodies
- (1)
- (1)
- (3)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (4)
- (1)
- (1)
- (3)
Latest Publications for our KCNQ4 Antibodies
: "Guanylyl Cyclase A/cGMP Signaling Slows Hidden, Age- and Acoustic Trauma-Induced Hearing Loss." in: Frontiers in aging neuroscience, Vol. 12, pp. 83, (2020) (PubMed).: "Hair cell maturation is differentially regulated along the tonotopic axis of the mammalian cochlea." in: The Journal of physiology, Vol. 598, Issue 1, pp. 151-170, (2020) (PubMed).
: "GC-B Deficient Mice With Axon Bifurcation Loss Exhibit Compromised Auditory Processing." in: Frontiers in neural circuits, Vol. 12, pp. 65, (2019) (PubMed).
: "Expression and function of Kv7.4 channels in rat cardiac mitochondria: possible targets for cardioprotection." in: Cardiovascular research, Vol. 110, Issue 1, pp. 40-50, (2016) (PubMed).
: "Role of Kv7 channels in responses of the pulmonary circulation to hypoxia." in: American journal of physiology. Lung cellular and molecular physiology, Vol. 308, Issue 1, pp. L48-57, (2015) (PubMed).
: "K(V)7.4 channels participate in the control of rodent renal vascular resting tone." in: Acta physiologica (Oxford, England), Vol. 214, Issue 3, pp. 402-14, (2015) (PubMed).
: "Kv 7 positive modulators reduce detrusor overactivity and increase bladder capacity in rats." in: Basic & clinical pharmacology & toxicology, Vol. 110, Issue 2, pp. 145-53, (2014) (PubMed).
: "KCNQ and KCNE potassium channel subunit expression in bovine retinal pigment epithelium." in: Experimental eye research, Vol. 116, pp. 424-32, (2014) (PubMed).
: "The endocannabinoid 2-AG controls skeletal muscle cell differentiation via CB1 receptor-dependent inhibition of Kv7 channels." in: Proceedings of the National Academy of Sciences of the United States of America, Vol. 111, Issue 24, pp. E2472-81, (2014) (PubMed).
: "Specification of skeletal muscle differentiation by repressor element-1 silencing transcription factor (REST)-regulated Kv7.4 potassium channels." in: Molecular biology of the cell, Vol. 24, Issue 3, pp. 274-84, (2013) (PubMed).
Aliases for KCNQ4 Antibodies
potassium voltage-gated channel subfamily Q member 4 (KCNQ4) Antibodiespotassium channel, voltage gated KQT-like subfamily Q, member 4 (kcnq4) Antibodies
potassium voltage-gated channel subfamily Q member 4 (Kcnq4) Antibodies
potassium voltage-gated channel, subfamily Q, member 4 (Kcnq4) Antibodies
DFNA2 Antibodies
DFNA2A Antibodies
k(v)7.4 Antibodies
KCNQ4 Antibodies
kv7.4 Antibodies
KV7.4 Antibodies
Did you look for something else?
- KCNQ3 Antibodies
- KCNQ2 Antibodies
- KCNQ1 Antibodies
- KCNN4 Antibodies
- KCNN3 Antibodies
- KCNN2 Antibodies
- KCNN1 Antibodies
- KCNMB4 Antibodies
- KCNMB3 Antibodies
- KCNMB2 Antibodies
- KCNMB1 Antibodies
- KCNMA1 Antibodies
- KCNK9 Antibodies
- KCNK7 Antibodies
- KCNK6 Antibodies
- KCNK4 Antibodies
- KCNK3 Antibodies
- KCNK2 Antibodies
- KCNK18 Antibodies
- KCNK17 Antibodies
- KCNQ5 Antibodies
- KCNRG Antibodies
- KCNS1 Antibodies
- KCNS2 Antibodies
- KCNS3 Antibodies
- KCNT1 Antibodies
- KCNT2 Antibodies
- KCNU1 Antibodies
- KCNV1 Antibodies
- KCNV2 Antibodies
- KCP Antibodies
- KCT2 Antibodies
- KCTD1 Antibodies
- KCTD10 Antibodies
- KCTD11 Antibodies
- KCTD12 Antibodies
- KCTD13 Antibodies
- KCTD15 Antibodies
- KCTD18 Antibodies
- KCTD4 Antibodies



